

引言 全球有超過3億人患有罕見病,已知的罕見病有7000多種,我國有2000多萬罕見病患者,每年新增患者超20萬,包括人們熟知的血友病、瓷娃娃、白化病、戈謝氏病、漸凍癥等。其中72%的罕見病是遺傳性的。 不幸的是,70%的患者在出生時或者兒童期已經發病。其中約30%的罕見病兒童壽命不超過15歲。更讓人揪心的是,罕見病種類繁多,臨床表現復雜多樣,導致診斷難、治療難。只有約5%的罕見病有相應的治療藥物(“孤兒藥”),超60%的患者甚至于無法得到明確診斷。 當前,除了藥物治療,干細胞臨床治療、酶替代療法和基因治療是罕見病研究領域的新焦點,也給罕見病患者帶來了新希望。 隨著科學研究的進展,這些新療法中有些已經開花結果,例如2018年美國批準了一款基因療法,用于治療發病率只有十萬分之二至三的罕見病Leber先天性黑蒙10型;再如,在過去幾年里,全球獲批上市的一些干細胞療法中,部分適應癥就是某個罕見病。

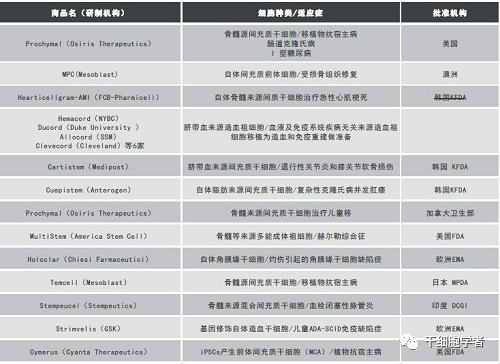
2009年,美國Osiris公司開發的人異基因骨髓來源間充質干細胞產品prochymal獲FDA批準上市,適應癥之一就是克羅恩病,另一個適應癥為兒童移植物抗宿主病。2012年這款干細胞療法獲加拿大批準上市,用于治療兒童移植物抗宿主病。
2012年,FDA批準了美國Athersys公司的干細胞療法 MultiStem?上市,適應癥為I型粘多糖貯積癥。
我國罕見病發展中心(CORD)發布的《中國罕見病參考目錄》中,粘多糖貯積癥和克羅恩病被列入目錄。
值得高興的是,當前在研的一些干細胞療法的適應癥正往罕見病方向延伸。例如,2016年在日本上市的針對移植物抗宿主病的骨髓間充質干細胞產品Temcell,今年正被拓展至大皰性表皮松解癥(EB)的治療研究中,而大皰性表皮松解癥也是我國《第一批罕見病》目錄中的一種罕見病。
此外,針對漸凍癥、帕金森等罕見病的干細胞療法陸續進入臨床試驗后期,距離上市越來越近。

在2018年上海市罕見病/孤兒藥學術年會上,中國工程院院士陳香美說道,“未來最有效的干細胞療法可能是在罕見病上。”
早衰癥又稱為科凱恩氏綜合征,是一種罕見遺傳病,患者身體衰老的速度比正常人快5-10倍,器官衰竭迅速,造成生理機能下降,目前臨床上尚無有效的治療方法。2016年,14歲早衰癥女孩桐桐成為了全球首位接受胎盤間充質干細胞治療的患者。
北京大學第三醫院在此前開展了一項臨床試驗,那也是胎盤干細胞治療早衰癥的全球首次嘗試。桐桐接受胎盤干細胞移植后,病情得到了緩解,皮膚變得比以前光滑,來了3次月經,聽力和肝功能有了改善的跡象。這則臨床研究案例充分展現了胎盤間充質干細胞治療早衰癥的前景。
除了早衰癥,國內在干細胞治療其他罕見病上也取得了喜人的結果。
2019年,復旦大學附屬兒科醫院使用干細胞移植治療兒科罕見病取得了突破,成功完成國內首例PIK3CD基因缺陷患兒的干細胞移植。

根據報道,自2014年起,復旦大學附屬兒科醫院已完成近200例兒童罕見病的干細胞移植治療,治療成功率為70-80%,挽救了很多常規藥物和手術治療無望的罕見病患兒,其中原發免疫缺陷病最多。
近年來,為了解決罕見病缺藥的問題,政府、企業以及公益界等多方合作共同發力。中共中央辦公廳、國務院辦公廳印發的《關于深化審評審批制度改革鼓勵藥品醫療器械創新的意見》,提出了鼓勵罕見病治療藥品醫療器械的一系列政策,特別指出,“罕見病治療藥品醫療器械注冊申請人可提出減免臨床試驗的申請。
對境外已批準上市的罕見病治療藥品醫療器械,可附帶條件批準上市,企業應制定風險管控計劃,按要求開展研究。”這意味著,罕見病新藥臨床試驗和審批時間有望縮短,盡快上市。
干細胞是一類具有自我復制能力的多潛能細胞,具有十分廣闊的應用前景,已被廣泛應用至多種疾病的臨床研究中,治療罕見病的研究案例也不再罕見,突破性成果不斷帶來新曙光,給治療罕見病帶來了很大的啟發。隨著干細胞新藥研發取得進展,罕見病的臨床治療有望迎來新的局面。
編輯:小果果,轉載請注明出處:http://www.448371.com/cells/gxb/37408.html
免責聲明:本站所轉載文章來源于其他平臺,主要目的在于分享行業相關知識,傳遞當前最新資訊。圖片、文章版權均屬于原作者所有,如有侵權,請及時告知,我們會在24小時內刪除相關信息。
說明:本站所發布的案例均摘錄于文獻,僅用于科普干細胞與再生醫學相關知識,不作為醫療建議。
 微信掃一掃
微信掃一掃  支付寶掃一掃
支付寶掃一掃 